
Por Mario Villada-Balbuena, Karol Desiree Lira Villanueva y Claudia Camacho-Zuñiga
El desarrollo y la aplicación de las vacunas contra Covid-19 fueron un hito en la ciencia médica. La tecnología tradicional basada en inocular virus debilitados para ocasionar una respuesta de nuestro sistema inmunitario, pero no la enfermedad, fue superada por otras más eficientes en el caso del SARS-CoV-2.
Como ya sabemos, las vacunas de Pfizer-BioNTech y Moderna utilizan Ácido Ribonucleico Mensajero (ARNm) para que las células produzcan la proteína de la espícula o spike, característica de la superficie del virus.
El Ácido Ribonucleico (ARN) es una de las macromoléculas básicas para la vida. Actúa como transportador de las instrucciones genéticas del Ácido Desoxirribonucleico (ADN) hacia los ribosomas (que son como fábrica de proteínas) y también juega un papel clave regulando cómo y cuándo se activan ciertos genes dentro de la célula.
El prefijo “macro” indica que está compuesto de entre 20 hasta miles de nucleótidos, donde cada nucleótido pesa entre 300 y 350 g/mol. Solo para dimensionar su tamaño y complejidad cabe recordar que una molécula de agua pesa sólo 18 g/mol.
Simular el comportamiento molecular
Hoy en día, se busca desarrollar tratamientos personalizados basados en ARN para ciertos tipos de cáncer, desórdenes genéticos o enfermedades infecciosas [1].
Las interacciones biológicas entre proteínas, enzimas, hormonas, antígenos, anticuerpos y nucleótidos no ocurren a través de reacciones tradicionales que forman enlaces químicos. Ocurren a través de fuerzas y acoplamientos de formas complementarias, formas tridimensionales que se ajustan entre ellas como en un rompecabezas, para activar diferentes funciones celulares.
La geometría tridimensional de las moléculas de ARN y las fuerzas requeridas para desdoblarlas se determinan experimentalmente utilizando instrumentos de alta precisión, costosos y complejos. Además, frecuentemente implican la destrucción de la muestra.
Desde este punto de vista, es muy valioso predecir la conformación espacial de tales moléculas y las fuerzas de interacción entre partículas.
Predecir la geometría y otras propiedades fisicoquímicas de las macromoléculas, antes de ser probadas en entes vivos o incluso, antes de ser sintetizadas, es el propósito de algunas simulaciones.
El uso de este tipo de simulaciones se ha incrementado con la potencia computacional. Los modelos más apegados a la evidencia experimental son muy valiosos en este campo. Sin embargo, en la predicción de la conformación de las macromoléculas, como proteínas, ADN o ARN, se requieren entre 1010 y 1018 iteraciones o “cálculos” [2].
Por esta razón, los modelos con requisitos computacionales más bajos también son altamente valorados.
Estrategia de simulación: «modelo de grano grueso»
El «modelo de grano grueso» es una estrategia para la simulación de macromoléculas, como el ARN, que simplifica su estructura al agrupar varios átomos en bloques más grandes.
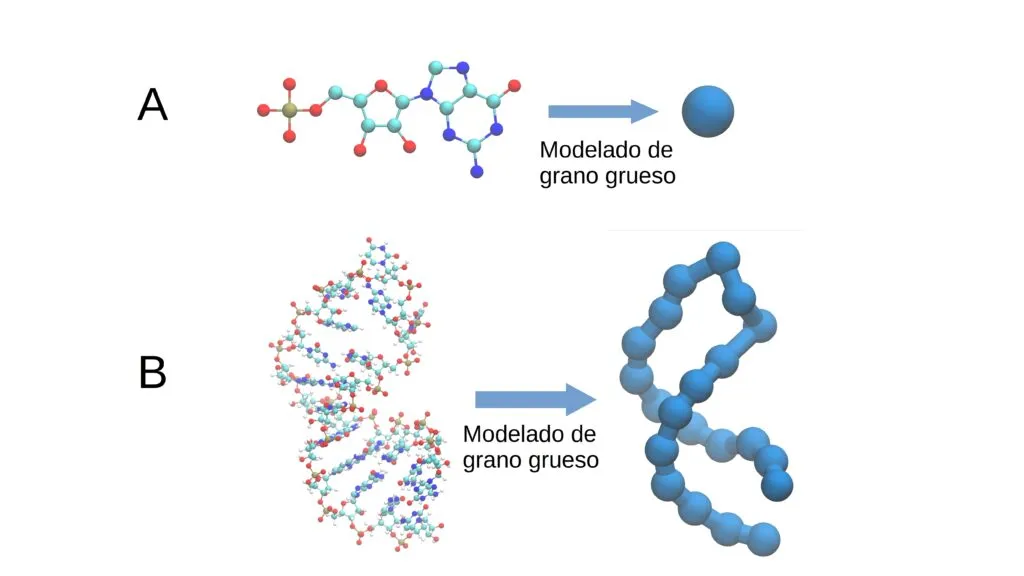
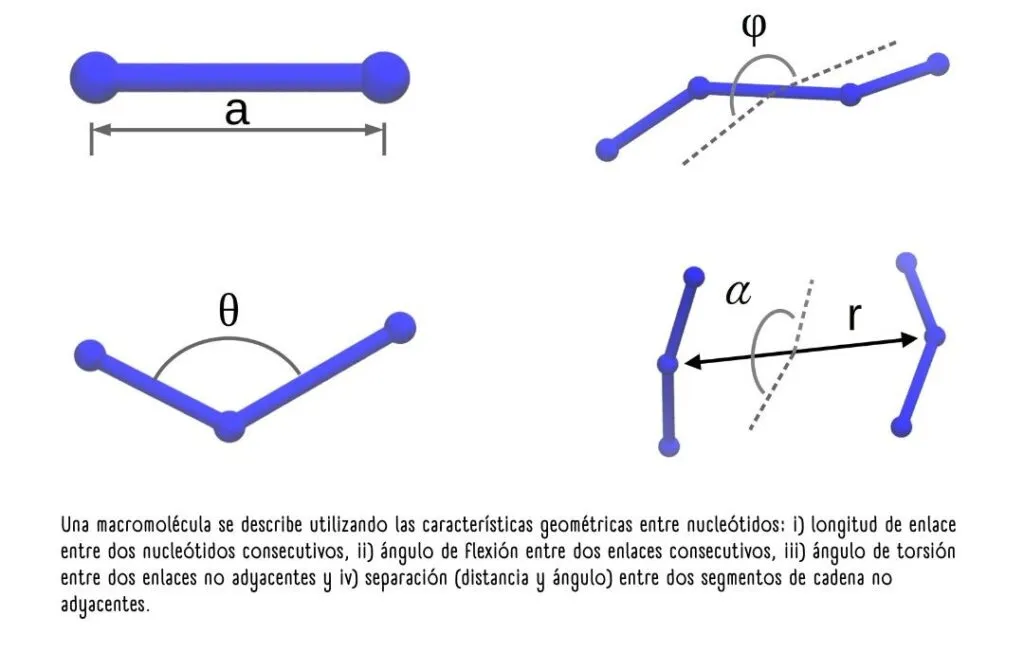
Esta figura(1) muestra la descripción de una molécula utilizando granos gruesos. Éstos agrupan varios átomos en un solo grupo o “grano”, permitiendo simplificar y eficientizar la simulación de procesos biológicos
En lugar de modelar la molécula átomo por átomo, ésta se representa con menos detalles, lo que reduce la complejidad y el tiempo de cálculo, sin sacrificar la precisión necesaria para entender su comportamiento. Este enfoque es crucial para estudiar procesos biológicos de manera eficiente con recursos computacionales limitados.
Un modelo de grano grueso simplifica la descripción de las moléculas. En arte, sería similar a un Van Gogh, donde se elimina el detalle para capturar las vibraciones más intensas en grandes pinceladas.
En otras palabras, un modelo de grano grueso describe la geometría de una macromolécula dando la posición no de uno de los miles de átomos que la forman, sino solo la posición de “granos” o grupos químicos de mayor tamaño.
En el caso del ARN, los granos pueden ser los nucleótidos mismos, formados por un grupo fosfato, una ribosa y una base nitrogenada (adenina, citosina, guanina o uracilo). Gracias a este proceso de simplificación, en vez de utilizar 60 variables para describirlos, se usaron solamente tres.
Explorando la dinámica del ARN
En el artículo One-bead coarse-grained model for RNA dynamics, se presenta un modelo de grano grueso de ARN partiendo de información experimental de estructuras tridimensionales disponibles en el Protein Data Bank [3]. Esta base de datos contiene las posiciones en tres dimensiones de cada uno de los átomos que forman a más de 220,000 moléculas.
Si bien los autores demuestran que esta simplificación logra ventajas de cálculo significativas, modelar la macromolécula sigue siendo un reto.
Aquí, aparece la belleza del enfoque cuántico. No es posible describir con exactitud la posición de una partícula, solamente describir la probabilidad de encontrarla en una cierta región del espacio.
Es entonces cuando se calculan las funciones de distribución de estos nucleótidos, o la probabilidad de encontrarlos en una conformación o acomodo geométrico específico.
Las distribuciones de probabilidad de las características de los nucleótidos permiten modelar la interacción energética entre ellas.
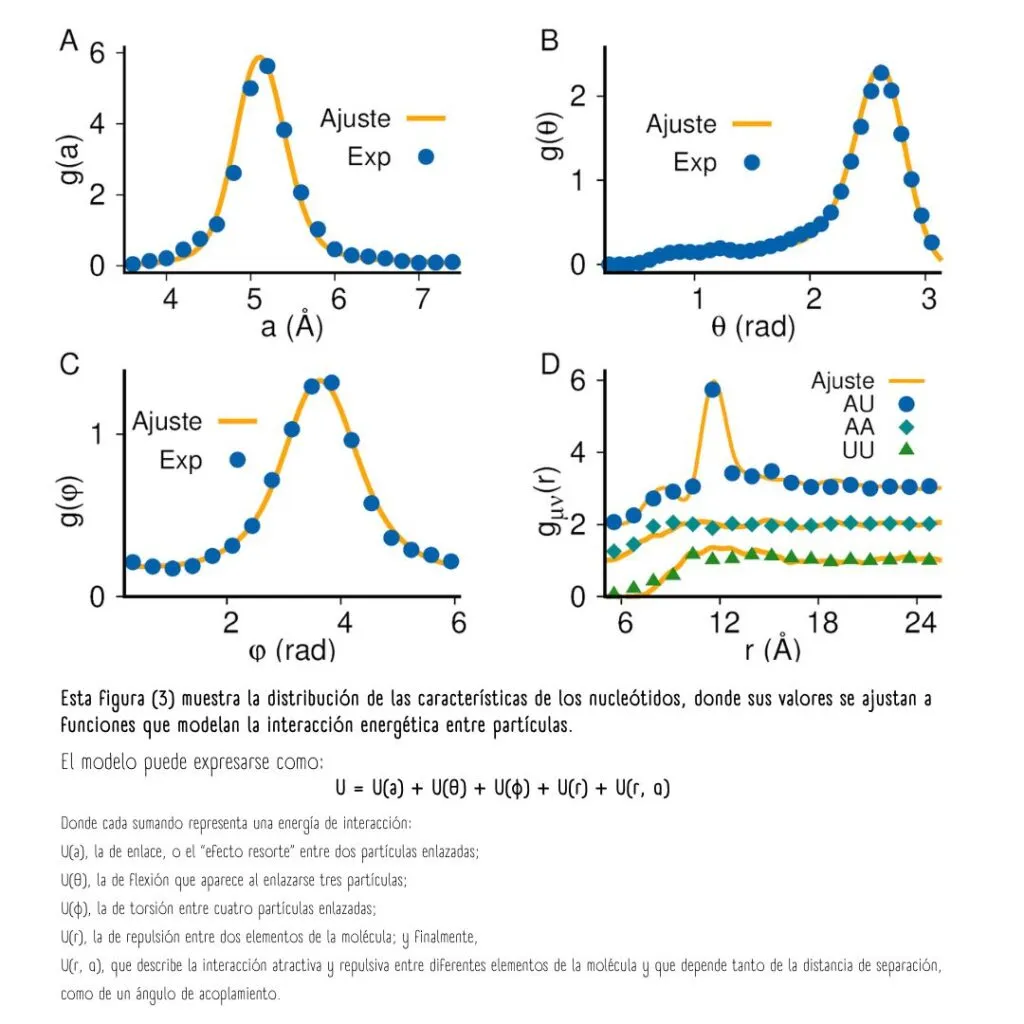
La suma de las energías entre todas las interacciones posibles representa la energía potencial total «U» de la cadena polimérica. «U» es el parámetro que debe optimizarse, tal como lo hace el Universo, porque los estados más estables son aquellos de menor energía.
Acomodo Tridimensional del ARN
Los avances tecnológicos han permitido modelar problemas complejos, como el estudio de las moléculas de ARN. A pesar de los progresos, aún existen desafíos sin resolver, como predecir su estructura tridimensional a partir de la secuencia de nucleótidos y entender cómo responden mecánicamente cuando se les aplica fuerza.
Imaginemos una cadena de ARN como una cuerda larga y delgada. Cuando en diferentes secciones de la cadena hay elementos que se atraen entre sí, estos se atraen formando una especie de horquilla o hairpins. Es decir, la atracción entre estas secciones constituyen un «doblador» en la cadena, dejando un pequeño bucle en el medio.
Un pseudonudo o pseudoknot es una estructura que a simple vista asemeja a un nudo. Sin embargo, no lo es, pues desaparece al estirar la estructura. En el estudio referido se predijo el acomodo tridimensional del ARN aplicando el modelo de grano grueso a 10 diferentes estructuras, incluidos hairpins y pseudoknots.
Para simular el acomodo de una cadena de ARN, en el estudio mencionado se inició colocando tres nucleótidos en posición aleatoria. Posteriormente, se simuló la interacción según las leyes de la física, hasta encontrar la configuración de menor energía. Este proceso se repitió hasta completar toda la secuencia de la molécula, determinando la estructura más estable. [4]
La conformación de moléculas de ARN simuladas (en azul) comparadas con las reportadas experimentalmente (en naranja) ¡parecen muy similares!
Sin embargo, en Física, como en otras ciencias, esta apariencia no es suficiente. Es necesario medirla. Para cuantificar dicha similitud, los autores calcularon diversas métricas de la semejanza estructural tanto global (dando un porcentaje respecto a toda la estructura), como local (considerando únicamente ciertas regiones de la molécula) [5].
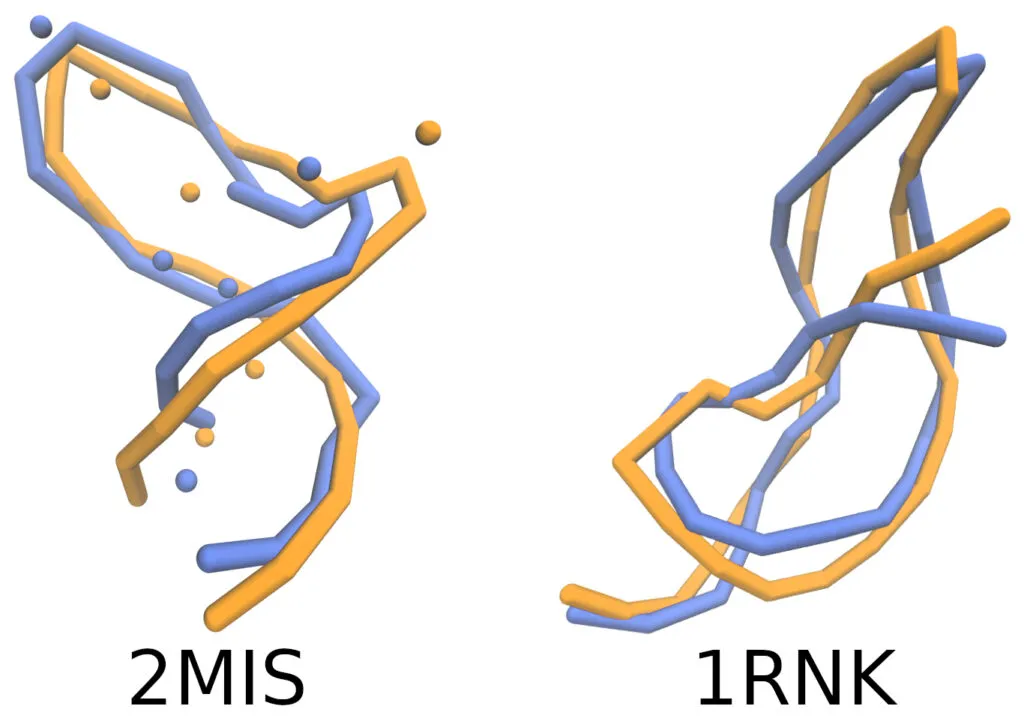
Los resultados cuantitativos evidenciaron una alta concordancia entre las simulaciones y los datos experimentales, a pesar de la simplicidad del modelo utilizado en comparación con otros modelos más complejos [6]. Una limitante es que a partir de la geometría final de la macromolécula en grano grueso es imposible reconstruir todos los detalles atómicos de la molécula.
Futuro en modelos de grano grueso
Estos avances son prometedores para el estudio de otras moléculas. De hecho, junto con Mauricio D. Carbajal-Tinoco también hemos simulado el desdoblamiento de algunas cadenas de ARN y continúamos mejorando el modelo y expandiendo la metodología a otras moléculas.
Aunque a nuestros ojos sean invisibles, las células son la unidad básica de los seres vivos. En este sentido, la comprensión del comportamiento del ARN y otras moléculas a nivel celular puede parecer ajena a nuestra vida cotidiana, hasta que se presentan problemáticas de salud global que requieren nuevas terapias y tratamientos médicos.
Esto significa que, los avances en la biotecnología y la comprensión de los procesos biológicos constituyen herramientas para mejorar la vida humana presente y futura.
Referencias
[1] Instituto Nacional del Cáncer. ¿Sirven las vacunas de ARN mensajero para tratar el cáncer?
[2] J. Jung, W. Nishima, M. Daniels, G. Bascom, C. Kobayashi, A. Adedoyin, M. E. Wall, A. Lappala, D. Phillips, W. Fischer, C. Tung, T. Schlick, Y. Sugita, y K. Y. Sanbonmatsu, Scaling molecular dynamics beyond 100,000 processor cores for large-scale biophysical simulations, J. Comput. Chem. 40, 1919 (2019).
[3] H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhati, H. Weissig, I. N. Shindyalov, y P. E. Bourne, Nucleic Acids Res. 28, 235 (2000).
[4] A. Iniesta y J. García de la Torre. A second‐order algorithm for the simulation of the Brownian dynamics of macromolecular models, J. Chem. Phys. 92, 2015 (1990).
[5] M. Villada-Balbuena y M. D. Carbajal-Tinoco. One-bead coarse-grained model for RNA dynamics, J. Chem. Phys. 146, 045101 (2017).
[6] C. J. Almeida, M.-F. Blanchet, M. Boniecki, J. M. Bujnicki, S.-J. Chen, S. Cao, R. Das, F. Ding, N. V. Dokholyan, F. S. Coulbourn, L. Huang, C. A. Lavender, V. Lisi, F. Major, K. Mikolajczak, D. J. Patel, A. Philips, T. Puton, J. Santalucia, F. Sijenyi, T. Hermann, K. Rother, M. Rother, A. Serganov, M. Skorupski, T. Soltysinski, P. Sripakdeevong, I. Tuszynska, K. M. Weeks, C. Waldsich, M. Wildauer, N. B. Leontis, y E. Westhof, RNA-Puzzles: A CASP-like evaluation of RNA three-dimensional structure prediction, RNA 18, 610 (2012).
.
Autores
Mario Villada-Balbuena. Doctor en Ciencias (Física) por el CINVESTAV-IPN y Físico por la UAEMex. Como posdoctorante en Biofísica Computacional, ha colaborado en proyectos de simulación con supercómputo financiados por CINVESTAV-IPN, UAEMéx y CONAHCYT. Miembro de la Red Temática de Materia Condensada Blanda del CONAHCYT, de la Biophysical Society (USA) y miembro de la RNA Society (USA). Profesor del departamento de Ciencias, Escuela de Ingeniería y Ciencias (EIC) del Tecnológico de Monterrey, Campus Toluca. También colabora en proyectos NOVUS de enseñanza de la ciencia y analítica de datos.
Karol Desiree Lira Villanueva. Estudiante de tercer semestre de ingeniería química en el Tecnológico de Monterrey, Campus Toluca.
Claudia Camacho-Zuñiga. Doctora en Ciencia de Materiales por la UAEMex, maestra en Ingeniería Química e Ingeniera Física por la Universidad Iberoamericana. Autora de cuatro libros de texto universitarios sobre Física y Matemáticas. Fundadora de Sistema Tlamatini para mejorar las habilidades de razonamiento matemático en niños y adolescentes. Investigadora SNI Nivel I. Actualmente, es investigadora en el Institute for the Future of Education y profesora en la Escuela de Ingeniería y Ciencias del Tecnológico de Monterrey, Campus Toluca, México.