
By Mario Villada-Balbuena, Karol Desiree Lira Villanueva y Claudia Camacho-Zuñiga
The development and deployment of COVID-19 vaccines marked a significant achievement in medical science. Traditional vaccine technology, which relies on injecting weakened viruses to trigger an immune response without causing disease, was outpaced by more efficient methods for SARS-CoV-2.
Pfizer-BioNTech and Moderna’s vaccines use messenger RNA (mRNA) to instruct cells to produce the spike protein, a signature feature on the virus’s surface.
Ribonucleic acid (RNA) is one of the fundamental macromolecules of life. It acts as a messenger, carrying genetic instructions from DNA to ribosomes — the protein factories of cells — and is crucial in regulating when and how specific genes are activated within a cell.
The prefix “macro” refers to the fact that RNA is made up of anywhere from 20 to thousands of nucleotides, each weighing between 300 and 350 g/mol. To put that into perspective, a single water molecule weighs just 18 g/mol.
Molecular Behavior Simulation
Today, RNA-based personalized treatments are being explored for certain cancers, genetic disorders, and infectious diseases.
Biological interactions between proteins, enzymes, hormones, antigens, antibodies, and nucleotides do not happen through traditional chemical bonding reactions. Instead, they rely on forces and complementary shapes — three-dimensional structures that fit together like puzzle pieces, triggering different cellular functions.
The 3D geometry of RNA molecules and the forces required to unfold them are determined through high-precision, costly, and complex experimental methods that often destroy the sample.
Given this, predicting the spatial structure of these molecules and the interactions between particles is incredibly valuable.
Simulations are used to predict the geometry and other physicochemical properties of macromolecules, sometimes even before they are tested in living organisms or synthesized. Advances in computational power have increased the use of these simulations. The most experimentally accurate models are highly valued, but predicting the conformation of macromolecules like proteins, DNA, or RNA requires between 10¹⁰ and 10¹⁸ iterations or “calculations.”
As a result, models with lower computational requirements are also highly prized.
Coarse-Grained Model
The “coarse-grained model” is a simulation strategy for macromolecules like RNA that simplifies their structure by grouping multiple atoms into larger blocks.
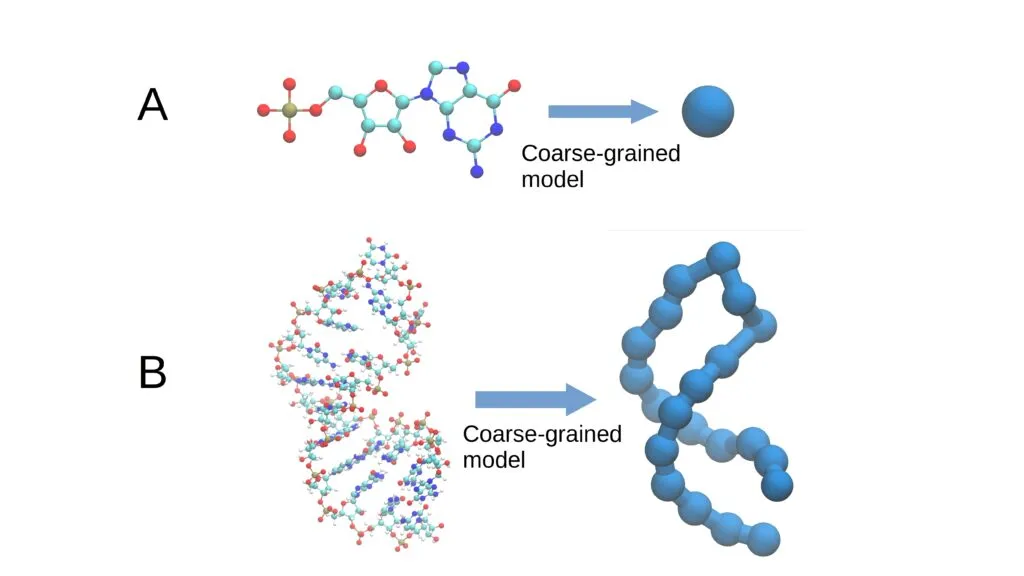
This figure (1) illustrates the description of a molecule using coarse grains. These group multiple atoms into a single cluster or “grain,” simplifying and streamlining the simulation of biological processes.
Rather than modeling a molecule atom by atom, this approach reduces detail, cutting complexity and computation time without sacrificing the accuracy needed to understand its behavior. This method is crucial for efficiently studying biological processes with limited computational resources.
A coarse-grained model simplifies molecular descriptions. In art, it’s akin to a Van Gogh painting, where fine details are stripped away to capture the essence through bold, sweeping strokes.
In essence, a coarse-grained model represents the geometry of a macromolecule by focusing not on the position of thousands of individual atoms, but on larger “grains” or chemical groups.
For RNA, these grains might be entire nucleotides, composed of a phosphate group, a ribose sugar, and a nitrogenous base (adenine, cytosine, guanine, or uracil). This simplification reduces the number of variables from 60 to just three.
Exploring RNA Dynamics
The article One-bead coarse-grained model for RNA dynamics introduces a coarse-grained RNA model based on experimental data from 3D structures available in the Protein Data Bank (PDB) [3], which contains the atomic positions of over 220,000 molecules.
While the authors demonstrate that this simplification offers significant computational advantages, modeling such a complex macromolecule remains a challenge.
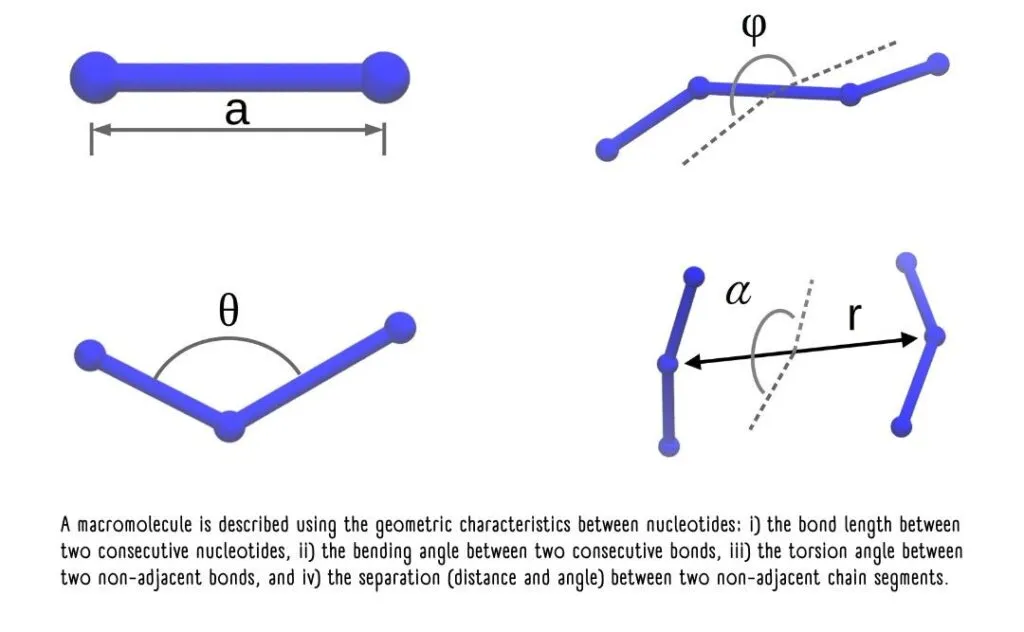
Here, the elegance of the quantum approach becomes apparent. It’s impossible to pinpoint the exact position of a particle; instead, we describe the probability of finding it within a certain region of space.
This is where the distribution functions of these nucleotides come into play, representing the likelihood of their being in a specific geometric conformation or arrangement.
The probability distributions of nucleotide characteristics enable the modeling of their energetic interactions.
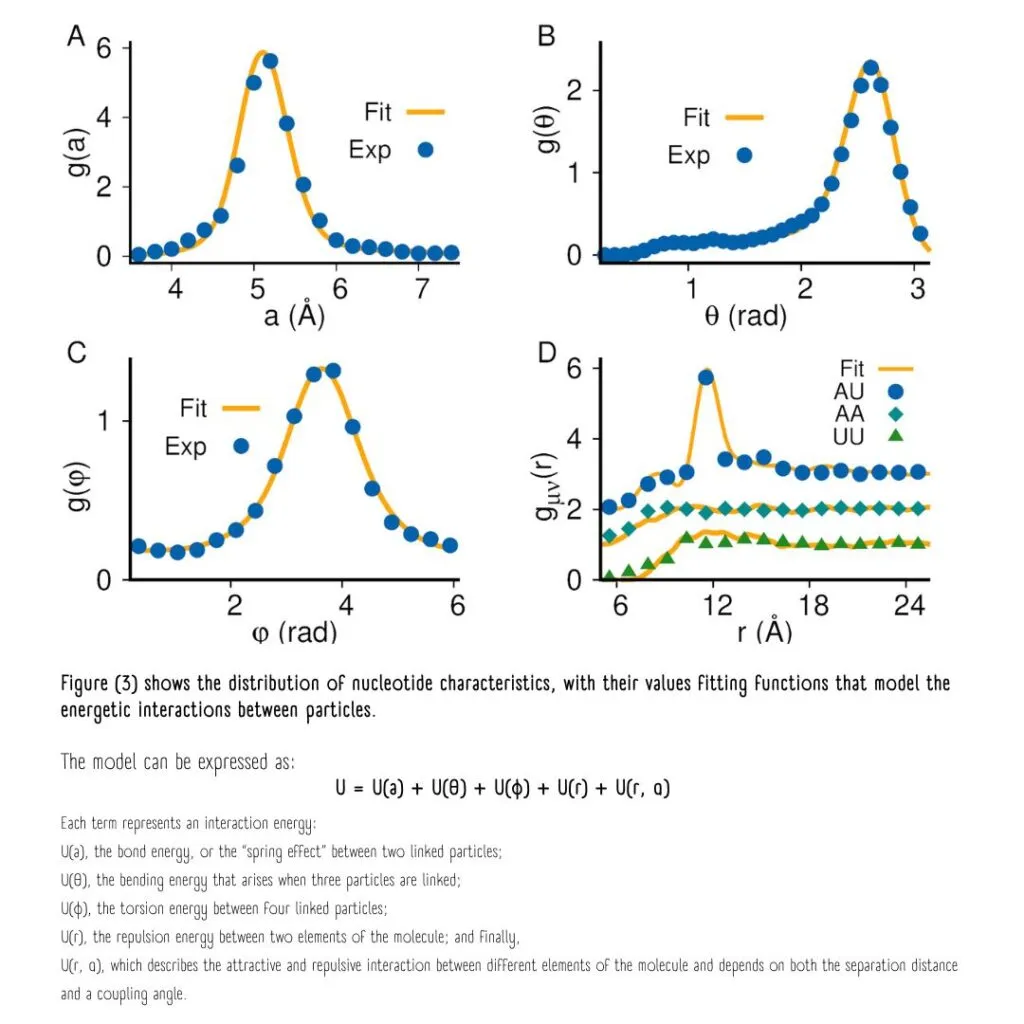
The sum of the energies from all possible interactions represents the total potential energy “U” of the polymer chain. “U” is the parameter that needs to be optimized, much like how the universe seeks to achieve states of minimum energy for stability.
Three-Dimensional Arrangement of RNA
Technological advances have enabled the modeling of complex problems, such as studying RNA molecules. Despite progress, challenges remain, such as predicting their three-dimensional structure from nucleotide sequences and understanding their mechanical responses to applied forces.
Imagine an RNA strand as a long, thin rope. When elements in different sections of the strand attract each other, they form structures similar to hairpins, creating loops in the chain.
A pseudoknot is a structure that superficially resembles a knot but disappears when the structure is stretched. The study in question predicted RNA’s three-dimensional arrangement by applying the coarse-grained model to 10 different structures, including hairpins and pseudoknots.
To simulate the arrangement of an RNA chain, the study began by randomly positioning three nucleotides. The interactions were then simulated according to physical laws until the configuration with the lowest energy was found. This process was repeated to complete the entire sequence, determining the most stable structure [4].
The simulated RNA conformations (in blue) compared to experimentally reported ones (in orange) appear strikingly similar!
However, in physics, as in other sciences, appearance alone is not sufficient. It must be measured. To quantify this similarity, the authors calculated various structural similarity metrics both globally (providing a percentage relative to the entire structure) and locally (focusing on specific regions of the molecule) [5].
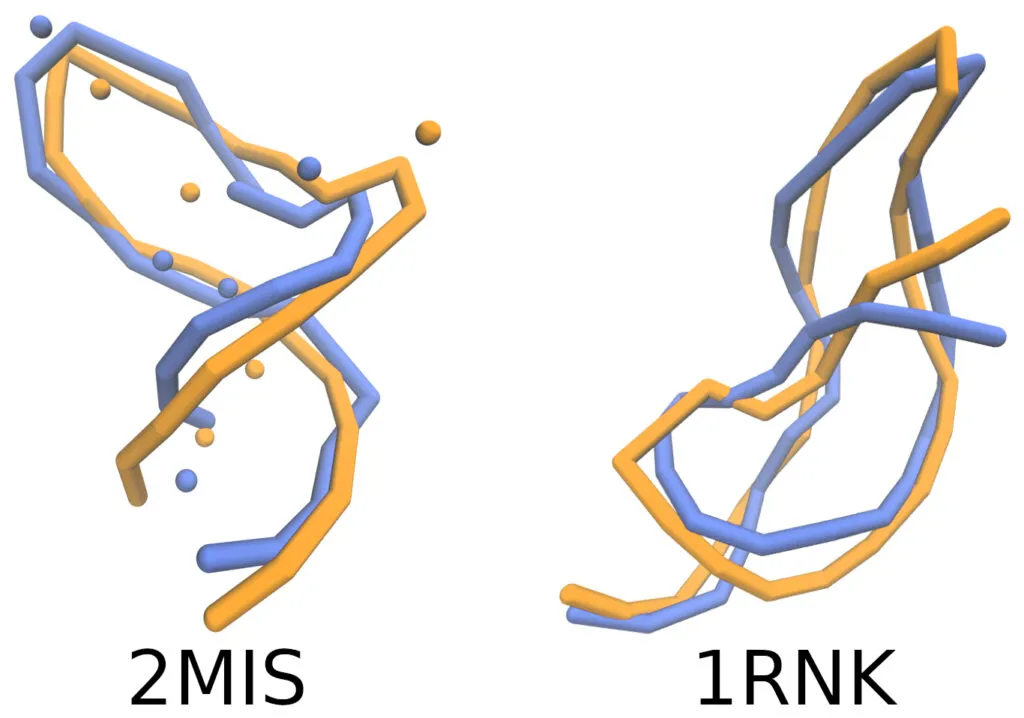
The quantitative results demonstrated a high degree of agreement between the simulations and experimental data, despite the simplicity of the model compared to more complex ones [6]. A limitation is that from the final coarse-grained geometry of the macromolecule, it is impossible to reconstruct all the atomic details of the molecule.
Future in Coarse-Grained Models
These advancements are promising for studying other molecules. In fact, alongside Mauricio D. Carbajal-Tinoco, we have also simulated the unfolding of certain RNA chains and continue to refine their model and expand their methodology to other molecules.
Although cells are invisible to the naked eye, they are the basic unit of life. In this sense, understanding RNA and other molecules at the cellular level might seem disconnected from our everyday lives until global health issues arise that require new therapies and medical treatments.
This underscores that advancements in biotechnology and the understanding of biological processes are crucial tools for saving and improving both present and future human life.
References
[1] Instituto Nacional del Cáncer. ¿Sirven las vacunas de ARN mensajero para tratar el cáncer?
[2] J. Jung, W. Nishima, M. Daniels, G. Bascom, C. Kobayashi, A. Adedoyin, M. E. Wall, A. Lappala, D. Phillips, W. Fischer, C. Tung, T. Schlick, Y. Sugita, y K. Y. Sanbonmatsu, Scaling molecular dynamics beyond 100,000 processor cores for large-scale biophysical simulations, J. Comput. Chem. 40, 1919 (2019).
[3] H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhati, H. Weissig, I. N. Shindyalov, y P. E. Bourne, Nucleic Acids Res. 28, 235 (2000).
[4] A. Iniesta y J. García de la Torre. A second‐order algorithm for the simulation of the Brownian dynamics of macromolecular models, J. Chem. Phys. 92, 2015 (1990).
[5] M. Villada-Balbuena y M. D. Carbajal-Tinoco. One-bead coarse-grained model for RNA dynamics, J. Chem. Phys. 146, 045101 (2017).
[6] C. J. Almeida, M.-F. Blanchet, M. Boniecki, J. M. Bujnicki, S.-J. Chen, S. Cao, R. Das, F. Ding, N. V. Dokholyan, F. S. Coulbourn, L. Huang, C. A. Lavender, V. Lisi, F. Major, K. Mikolajczak, D. J. Patel, A. Philips, T. Puton, J. Santalucia, F. Sijenyi, T. Hermann, K. Rother, M. Rother, A. Serganov, M. Skorupski, T. Soltysinski, P. Sripakdeevong, I. Tuszynska, K. M. Weeks, C. Waldsich, M. Wildauer, N. B. Leontis, y E. Westhof, RNA-Puzzles: A CASP-like evaluation of RNA three-dimensional structure prediction, RNA 18, 610 (2012).
.
Authors
Mario Villada-Balbuena. Ph.D. in Science (Physics) from CINVESTAV-IPN and Physicist from UAEMex. As a postdoctoral researcher in Computational Biophysics, he has collaborated on supercomputing simulation projects funded by CINVESTAV-IPN, UAEMex, and CONACYT. He is a member of the CONACYT Soft Condensed Matter Network, the Biophysical Society (USA), and the RNA Society (USA). He is a professor in the Department of Sciences at the School of Engineering and Sciences (EIC) at Tecnológico de Monterrey, Toluca Campus. He also works on NOVUS projects for science education and data analytics.
Karol Desiree Lira Villanueva. Third-semester chemical engineering student at Tecnológico de Monterrey, Toluca Campus.
Claudia Camacho-Zuñiga. Ph.D. in Materials Science from UAEMex, M.Sc. in Chemical Engineering, and B.Sc. in Physics Engineering from Universidad Iberoamericana. Author of four university textbooks on Physics and Mathematics. Founder of Sistema Tlamatini, which aims to improve mathematical reasoning skills in children and adolescents. SNI Level I researcher. Currently, she is a researcher at the Institute for the Future of Education and a professor at the School of Engineering and Sciences at Tecnológico de Monterrey, Toluca Campus, Mexico.
.